本资料根据浙江大学黄方亮老师(huangfl@zju.edu.cn)、冯晔老师(pandafengye@zju.edu.cn)的课程 ppt 等整理。
未经两位老师和我本人许可,请勿以任何方式转载或引用本文。
概论
基本概念
生物芯片(biochip)指通过机器人自动印迹或光引导化学合成技术在硅片、玻璃、凝胶或尼龙膜上制造的生物分子微阵列,根据分子间的特异性相互作用的原理,将生命科学领域中不连续的分析过程集成于芯片表面,以实现对细胞、蛋白质、基因及其他生物组分的准确、快速、大信息量的检测。
基因芯片发展历史
- Southern & Northern Blot
- Dot Blot
- Macroarray
Microarray
- 1991年Affymatrix公司福德(Fodor)组织半导体专家和分子生物学专家共同研制出利用光蚀刻光导合成多肽;
- 1992年运用半导体照相平板技术,对原位合成制备的DNA芯片作了首次报道,这是世界上第一块基因芯片;
- 1993年设计了一种寡核苷酸生物芯片;
- 1994年又提出用光导合成的寡核苷酸芯片进行DNA序列快速分析;
- 1995年,斯坦福大学布朗(P.Brown)实验室发明了第一块以玻璃为载体的基因微矩阵芯片;
- 1996年灵活运用了照相平板印刷、计算机、半导体、激光共聚焦扫描、寡核苷酸合成及荧光标记探针杂交等多学科技术创造了世界上第一块商业化的生物芯片;
- 2001年,全世界生物芯片市场已达170亿美元,用生物芯片进行药理遗传学和药理基因组学研究所涉及的世界药物市场每年约1800亿美元。
基因芯片技术的特点
- 高度并行性:提高实验进程、利于显示图谱的快速对照和阅读。
- 多样性:可进行样品的多方面分析,提高精确性,减少误差。
- 微型化:减少试剂用量和反应液体积,提高样品浓度和反应速率。
- 自动化:降低成本,保证质量而且灵敏度高。
生物芯片的分类
一、按载体材料分类
- 玻璃芯片
- 硅芯片
- 陶瓷芯片
二、点样方式分类
- 原位合成芯片:采用显微 光 蚀刻等技术在特定部位原位合成寡核苷酸而制备的芯片。探针较短。
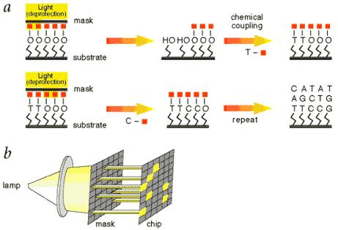
- 微矩阵芯片:将预先制备的DNA片段以显微 打印 的方式有序地固化于支持物表面而制成的芯片。探针的来源较灵活。
- 电定位芯片:利用 静电 吸附的原理将DNA快速定位在硅基质、导电玻璃上,在电力推动下进行快速杂交。
三、按生物类型分类
- 基因芯片或DNA芯片(Genechip):是根据 核酸杂交 的原理,将大量 探针分子 固定于支持物上,然后与标记的样品进行杂交,通过检测杂交信号的强度及分布来进行分析。
- 蛋白质芯片(proteinchip):利用 抗体与抗原 特异性结合即 免疫 反应的原理,将蛋白质分子(抗原或抗体)结合到固相支持物上,形成蛋白质微阵列,实现抗原抗体的互检,即蛋白质的检测。
- 细胞芯片(cellchip):是将细胞按照特定的方式固定在载体上,用来检测 细胞间相互影响 或相互作用。
- 组织芯片(tissuechip):是将组织切片等按照特定的方式固定在载体上,用来进行免疫组织化学等组织内 成分差异 研究。
- 芯片实验室:微流控芯片,lab-on-chip 采用纳米技术,制造出微小结构:变性、分离、纯化、电泳、PCR扩增、加样及检测,使各个实验步骤微缩于一个芯片上。高度集成化的集样品制备、基因扩增、核酸标记及检测为一体的便携式生物分析系统。实现生化分析全过程集成在一片芯片上完成,从而使生物分析过程自动化、连续化和微缩化。芯片实验室是生物芯片技术发展的最终目标。

测序技术简史
一代DNA测序历史
- 70年代末,WalterGilbert发明化学法、FrederickSanger发明双脱氧终止法手动测序,同位素标记
- 80年代中期,出现自动测序仪(应用双脱氧终止法原理)、荧光代替同位素,计算机图象识别
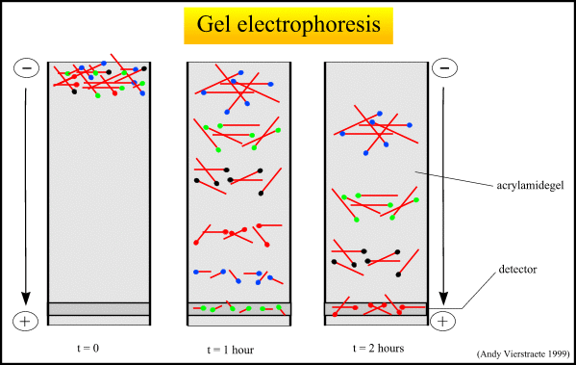
- 90年代中期,测序仪重大改进、集束化的毛细管电泳代替凝胶电泳
- 2001年完成人类基因组框架图

一代测序仪 MegaBACE1000
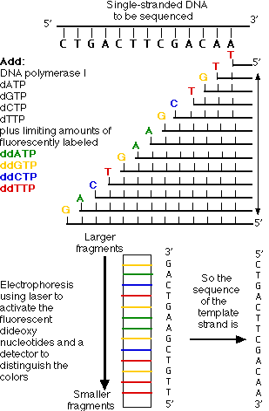
双脱氧终止法测序反应原理
原料
- DNA聚合酶
- DNA模板
- 单链寡核苷酸引物
- 4 种 dNTP(dATP、dGTP、dTTP和dCTP)
4 种 ddNTP(ddATP、ddGTP、ddTTP和ddCTP)
DNA聚合酶在模板指导下,不断地逐个将dNTP加到引物的3’-OH末端,使引物延伸,合成出新的与模板互补DNA链。在链的延长过程中一旦加入双脱氧核苷三磷酸(ddNTP),由于其双脱氧核糖的3’位置缺少一个羟基,不能同后续的dNTP形成磷酸二酯键,即形成一系列具有相同5’-引物端和以ddN残基为3’端结尾的 长短不一的片段的混合物,通过毛细管电泳并经过分析从而获得模板DNA的核苷酸序列。
Illumina 测序原理
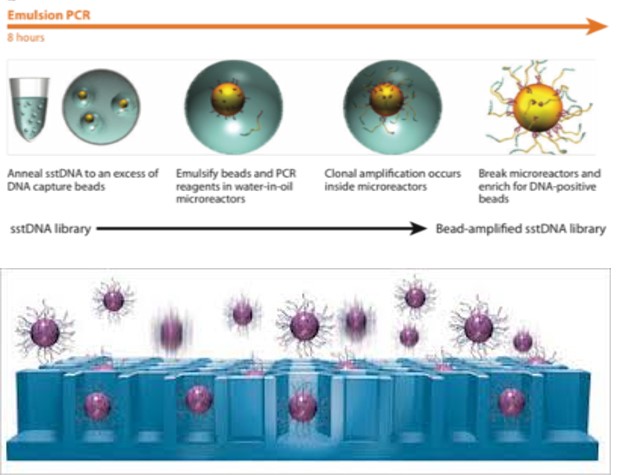
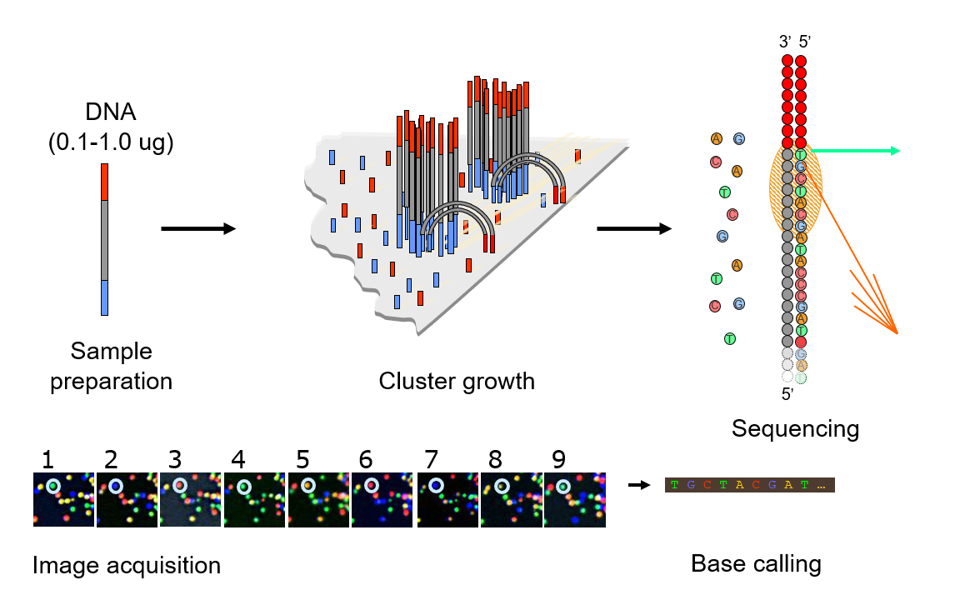
二代测序仪功能
- 未知及已知物种 表达谱, 基因组测序
- 鉴定 RNA 可变剪切
- 环境 (宏)基因组 (Metagenomics)测序
- 不同PCR产物 混合测序
- DNA 甲基化 鉴定
- 小RNA,非编码RNA 测序
- 染色质 免疫共沉淀分析(chip-seq)
- 病症筛查,唐氏综合症筛查,癌症用药基因敏感位点筛查,新生儿疾病相关基因位点筛查

二代测序数据格式
- Fastq,Fasta
- Bam
- Sam
- Tiff
当二代测序的原始数据拿到手之后,第一步要做的就是看一看原始reads的质量。常用的工具就是fastqc(使用方法见第三章PPT)
fastq
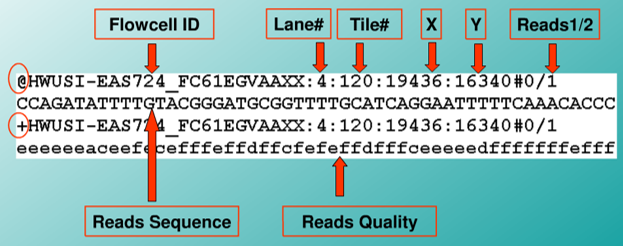
打分公式
SAM,BAM格式
SAM是一种 序列比对格式标准 ,由sanger制定,是以TAB为分割符的文本格式。主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果。当测序得到的fastq文件map到基因组之后,我们通常会得到一个sam或者bam为扩展名的文件。SAM的全称是sequence alignment/map format。
SAM和BAM是 序列比对 之后常用的输出格式,比如tophat输出BAM格式,bowtie和bwa等都采用了SAM格式。
BAM格式其实就是SAM格式的 二进制格式,占用存储空间更小。
在SAM输出的结果中每一行都包括十二项通过Tab分隔,从左到右分别是:
- 序列的名字
- 概括出一个合适的标记,各个数字分别代表
- 参考序列的名字
- 在参考序列上的位置
- mapping quality 越高则位点越独特
- 代表比对结果的CIGAR字符串,
- mate 序列所在参考序列的名称
- mate 序列在参考序列上的位置
- 估计出的片段的长度,当mate 序列位于本序列上游时该值为负值。
- read的序列
- ASCII码格式的序列质量
- 可选的区域
第三代测序技术

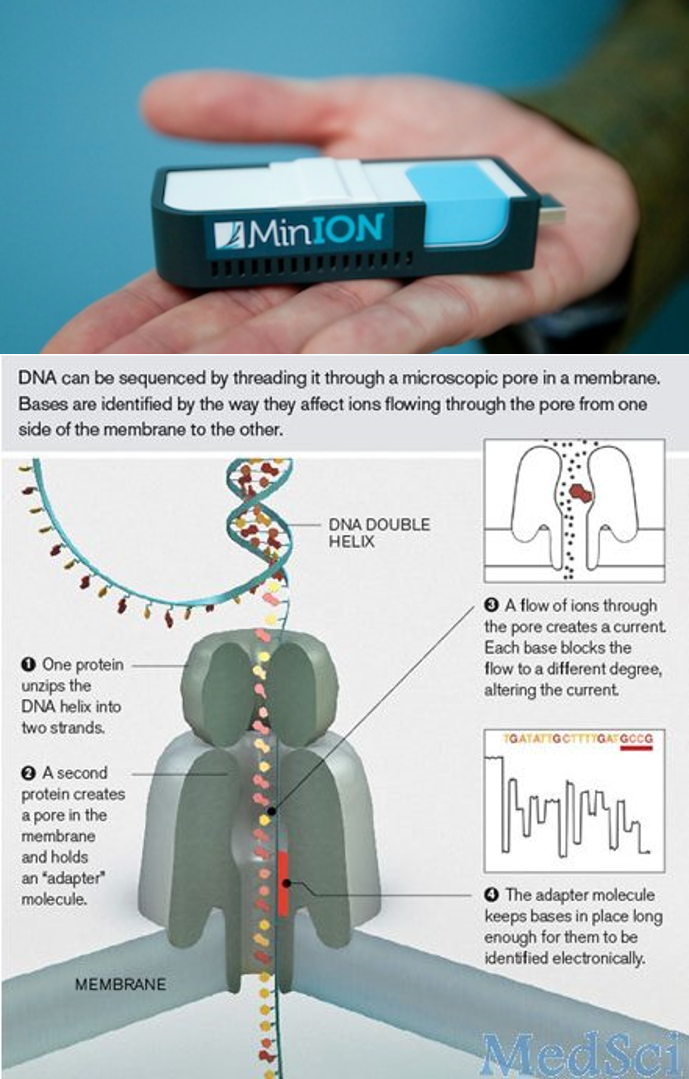
纳米孔测序仪,错误率36%,120Kb
DNA芯片技术的操作流程
芯片的制作准备
- 点阵设计(芯片上核酸探针序列的选择以及排布)
- 固定在芯片上的生物分子样品
- 固相支持物(即芯片片基)
- 制作芯片的仪器
固相支持物的种类及预处理
考虑荧光背景的大小、化学稳定性、结构复杂性、介质对化学修饰作用的反应、介质表面积及其承载能力以及非特异吸附的程度等因素。
芯片制备方法
- 原位合成(in situ synthesis)适用于:寡核苷酸
- ① 光导原位合成法
- ② 原位喷印合成
- ③ 分子印章多次压印合成
- 点样法 适用于:大片段DNA、寡核苷酸、mRNA
- ① 接触式点样(针式打印)
- ② 非接触式点样(喷墨打印)
原位合成法(in situ synthesis)
又可分为原位光控合成法和原位标准试剂合成法。适用于寡核苷酸,使用 光引导 化学原位合成技术。是目前制造高密度寡核苷酸最为成功的方法。
优势:合成速度快、步骤少、探针数目呈指数增长、阵列密度高;
缺陷:但光蚀刻掩膜昂贵、每步产率较低,因此合成探针长度较短。
- 测序、查明点突变;
- 高密度、根据已知的DNA编制程序;
- 制作复杂、价格昂贵、不能测定未知DNA序列。
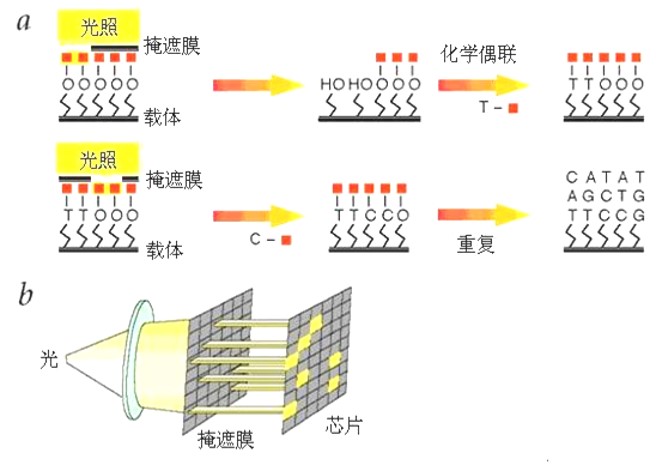
光敏保护基团;光蚀刻掩膜(或蔽光膜)
压电打印法
类似彩色喷墨打印,但有多个芯片喷印头及储液囊,储液囊中装四种碱基合成试剂,喷印头在整个芯片上移动。
- 其化学原理:固相合成 DNA
- 40~50nt,产率较高
点样法
- 设计点阵
- 人工合成的寡核苷酸片段;
- PCR、RT/PCR 等扩增的 DNA 或 cDNA 片段;
- 基因组提取的 DNA 片段;
- 表面带正电荷的芯片支持物
- 紫外交联固定、Schiff碱连接法固定

待检测样品的制备和标记
- 待检测样品中 mRNA 或 DNA 的提取及纯化
- RNA的稳定性
- 液氮或干冰; 立即抽提
- RNA 保护剂
- 纯化
- RNA的稳定性
- RT-PCR 或 PCR 扩增靶片段
- 固相PCR系统
- 样品的标记
- 伴随RT or PCR过程
- 随机引物法、缺口平移法等
- 标记物
- 荧光染料,如Cy3、Cy5
- 生物素、地高辛
- 放射性核素,如 32P、33P
- 化学发光
- 金属离子Au和Ag(纳米金属微粒探针)
- 双/多色荧光标记
- 标记产物纯化
检测表达量的步骤
- mRNA提取
- cDNA合成、PCR
- 标记
SNP或者突变检测、Genotyping
- Genomic DNA 纯化
- 标记
一旦生物样品被收集分离,它的 RNA 会立刻变得 非常不稳定,极易被降解。由于特异及非特异的RNA降解或者由于应激反应产生新的RNA都会引起RNA状态的改变。因此,采样后需立即稳定样品里的RNA以保存当时RNA的表达状态。
将液氮或干冰带到采样现场,采样后立即抽提RNA或者运回实验室保存。
RNAlater RNA Stabilization Reagent, 只要在采样后立即将新鲜样品浸入这种液体试剂,RNA保护剂可以迅速渗透到组织或其他生物样本中,稳定并保护RNA完整而不被降解,确保下游分析得到的数据真实反应样品的表达信息。37度1天;18~25度7天,2-8度4周,-20度永久保存。特别适用于动物组织(切成<0.5cm),以使迅速渗透、培养细胞、细菌、白细胞,不适用全血或体液。
纯化柱
标记物要注意适用的芯片片基,如有的化学发光试剂盒不适用于玻片基质;间接荧光标记适用玻片基质。
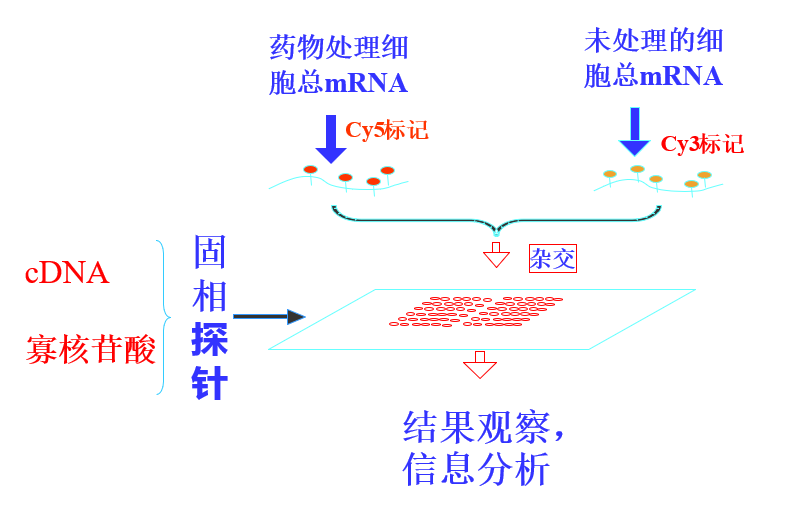

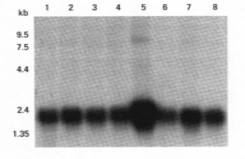
Cy3、Cy5双色荧光标记结果示意图
分子杂交
配套商业化芯片有配套杂交试剂、杂交盒;自制芯片,可自行配,也可购买杂交buf,洗膜液,有不同严谨程度的杂交液可供选择。毕竟在芯片这样精密的实验中,在这么小的一个点上的DNA的两是非常有限的,要得到理想的结果需要非常精确的反应条件。
- 芯片杂交盒、洗片器;芯片杂交仪
- 杂交液
- 清洗液
- 影响杂交的因素
- 时间、温度、缓冲液等,
- 根据芯片上核酸片段的长短、用途选择杂交条件
DNA 条形码
条形码技术是应现代零售业发展的需求而产生的, 在零售业的商品管理与销售中发挥了无法替代的关键作用, 生物分类学家从中得到了启示:DNA分子一级结构上的线性核苷酸排列可以建立类似的生物条形码,应用于快速鉴别生物。
DNA 条形码的理想序列有3 个基本判断标准:
- 序列变异水平适宜,可以将不同物种彼此区分开来,同时种内变异较小;
- 变异区域两端的序列高度保守,可以设计在众多物种中稳定扩增的通用引物;
- 扩增序列尽量短,最好一个反应可以完成测序.
DNA条形码技术的分析流程
- 采集所研究样品,记录采集时间和地点,并利用酒精或冷冻保存样品,采用适宜方法提取DNA;
- 利用相关引物对目的片段进行PCR扩增;
- 对扩增片段进行测序;
- 利用相关软件校正序列,建立系统进化树,并分析结果;
- 将研究样品的DNA序列,图像,采集地点和日期,采集人和鉴定人等信息提交DNA条形码数据库。
基因芯片的应用
基因表达分析
针对基因的 保守区段 设计多对 完全匹配的寡核苷酸探针(PM) 和与之相应的 中心单碱基错配的寡核苷酸探针(MM),固定于芯片的 相邻位置 上,与标记的样品靶序列杂交。正常完全匹配的情况下(阳性),PM的杂交信号明显强于MM的杂交信号,而错配时(假阳性),两者的杂交信号区别不明显,(PM/MM)信号的比值可作为衡量阳性的指标。针对某一基因的三条以上探针呈阳性时,可定性的判定基因的表达。而通过比较正常和异常样品杂交信号的差异,来检测不同样品中基因表达水平的变化。
基因功能分析研究
将成千上万个我们克隆到的特异性靶基因固定在一块芯片上,对来源于不同情况细胞的mRNA或逆转录所得的cDNA进行检测,从而对这些基因表达的个体特异性、组织特异性、发育阶段特异性、分化阶段特异性等进行综合评定与判断。
疾病的基因诊断
目前诊断芯片主要涉及:
- 癌症
- 心血管疾病
- 血液病
- 遗传性疾病
- 神经系统疾病
- 部分感染性疾病
- 免疫反应相关性疾病
- 毒物引起的损伤等
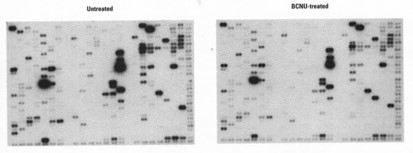
药物筛选
经过基因功能研究,可以确立与某些疾病相关的基因以及它们的表达情况。将这些基因特异性片段固定在芯片上,研究病变组织和正常组织在某些药物刺激下这些基因表达的变化情况,可快速判断药物作用的效果,并进行高通量筛选。
检测基因突变
根据基因突变热点所有可能的突变情况设计多条寡核苷酸探针,合成、固定在芯片上的特定位置,与PCR扩增、标记的靶基因相应区段杂交,根据特定位置上杂交信号的有无和与之相应的探针序列来判定基因突变的类型。
- RNA 的检测:表达丰度,剪切体
- DNA 的检测: SNP , CGH , Methylation
- cDNA芯片:表达谱芯片, CGH
- 寡核苷酸芯片:诊断芯片,SNP分型芯片
探针设计原则
以已知序列的突变热点位置为中心,按从左到右的顺序在目标序列中取一定长度的互补片段(<25bp)作为探针。对于这组探针,其中之一为野生型,而其他三种为突变型探针。将此四组探针顺序地点样于芯片上相邻的四行,分别为A、G、C、T行,同时与扩增得到的标记的易变基因区段DNA杂交,根据杂交信号的位置可确定突变的类型。
- 探针序列应为特异性强和灵敏度高的寡核苷酸。
- (G+C)% 应在 40~60% 。
- 避免内部“发卡”结构 (连续互补 < 4 nt)。
- 避免同一碱基的连续出现 ( < 4 nt)。
- 探针与其他已知的各种基因序列进行同源性比较,若此探针序列与 非靶基因 序列有70%以上的同源性或连续8个以上的碱基序列相同,则最好不用。
- 设置多个序列相近的参考寡核苷酸探针来检测同一基因。
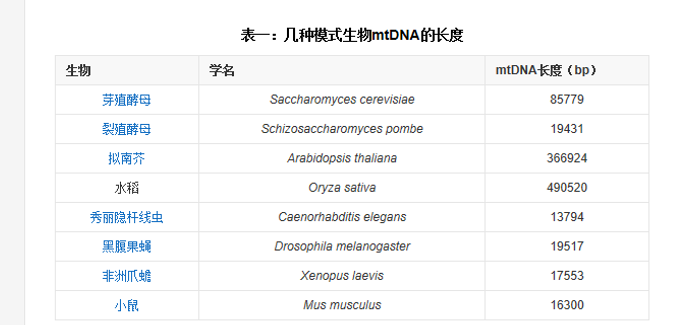
线粒体和叶绿体 DNA
线粒体
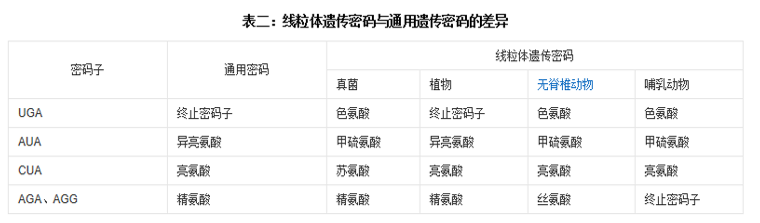
mtDNA 和 核 DNA 的主要差异:
- 长度
- 密码子对应氨基酸
- mtDNA 几乎不发生基因重组(群体遗传学、进化生物学)
叶绿体
- 双链环状
- 约含12个cpDNA分子
- 大小在120kb到217kb之间,相当于噬菌体基因组的大小
- 编码与光合作用密切相关的一些蛋白和一些核糖体蛋
- 基因表达调控是在不同水平上进行的,光和细胞分裂素对叶绿体基因的表达也起着重要的调节作用
- 基因组由两个反向重复序列(IR)和一个短单拷贝序列(short single copy seguence, SSC)及一个长单拷贝序列(long single copy seguence, LSC)组成
叶绿体基因组DNA检测
- 采集没有检测过叶绿体基因组的物种样本
- 提取物种总DNA
- 根据基因条形码技术(rbcl基因)确认物种
- 酶解法富集叶绿体DNA
- 构建高通量测序文库:
- 纯化富集后的叶绿体DNA
- 检测浓度,取 50ng 构建高通量测序文库
- 酶切法构建测序文库
操作流程
- 利用 DOGMA 或 CPGAVA S软件 注释 叶绿体基因组中的基因
- 利用 tRNAscan-SE 1.21 软件确定 tRNA 基因的边界。注释结果利用 Apollo 软件进行 修订, 调节编码基因 起始终止位置。
- 利用 sequin 软件完成五味子叶绿体基因组的 提交。
- 利用 OGDRAW 软件画出五味子叶绿体基因组 环形图谱。
- 利用 mVISTA 软件, 在 Shuffle-LAGAN 模型下, 将五味子叶绿体基因组与其他5种基部被子植物 (少药八角、白睡莲(Nymphaea alba L.)、Trithuria inconspicua Cheeseman、Amborella trichopoda Baill. 和Nuphar advena (Aiton) W.T. Aiton) 叶绿体基因组进行 比较分析。
- 利用 MISA 软件 检测叶绿体基因组 简单重复序列(simple sequence repeats, SSR)。
- Tandem repeats finder v4.04 软件被用于检测 串联重复(tandem, T), 参数默认。
分析结论示例
研究完成了五味子叶绿体基因组 测序、组装与注释,并对其 结构、GC含量、密码子使用率、重复序列 等进行了分析。结果显示,五味子叶绿体基因组结构和基因组成与大多数被子植物类似,但 ycf15 基因缺失。与大多数被子植物相比,五味子叶绿体基因组的 IR 区发生了约 10 kb 的收缩。通过叶绿体基因组系统进化分析,五味子的 系统发育位置 得到确认。五味子作为传统药用植物,具有重要药用和经济价值,其叶绿体基因组的测序与分析,为五味子科 系统进化分析 和 物种鉴定 提供了数据基础,并为解决近缘种进化关系和物种鉴定等提供了新的方法。
软件和数据库
网络设计引物软件
PRIMER3
看测序图软件
chromas
bioedit
一代测序序列分析软件DNASTAR
SEQMAN
EDITSEQ
基因组拼接
CLC Genomics Workbench
适合于NGS数据的基因组组装软件
- ALLPATHS-LG
- Velvet
- SOAPdenovo
- Bambus2
- CABOG
- MSR-CA
- SGA
- VCAKE
- SHARCGS
- SSAKE
- Euler
基因组注释
微生物基因组注释和基因预测工具:Glimmer
直接用SEQIN软件上传到NCBI,官方注释。
比较基因组数据库
TIGR(The Institute of Genomic Research Database-TDB,美国基因组研究所的数据库)是国际上重要的测序中心之一,其重点是假设的基因组学和比较基因组的研究。它有大量的基因组数据和标记表达序列数据,其资源以微生物为主,真核生物和人类基因组为辅。TIGR数据库包括了微生物、植物及人类的DNA及蛋白质序列, 基因表达, 细胞的作用, 蛋白质家族及分类数据,是一套大型综合数据库。在它收录的多种多样的数据库中,微生物基因组数据库是世界上著名的基因组数据库。,此外,其还拥有世界上最大的cDNA数据库。由该页面可进入以下数据库:微生物库(MDB),人类基因索引,老鼠基因索引,水稻(Rice)基因索引,人类基因组排序项目,人类cDNA图项目,表达的基因结构库等。
基因组拼接
基本概念
- Read:一条测序序列即为一条read;
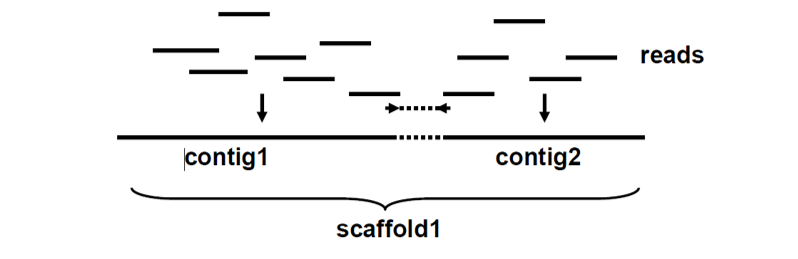
- Contig:重叠群,拼接一致序列片段,中间没有gap;
- Scaffold:框架序列,基于文库正反向信息确定的contigs顺序框架,中间可能有gap;
- N50:将各个序列按长度大小排列,从大到小扫描累加长度值,累加值首次超过所有序列总长的50%时,扫描到的该序列长度为N50。
De novo、Re-sequencing
从头测序之前没人做过,重测序可以用来找SNP。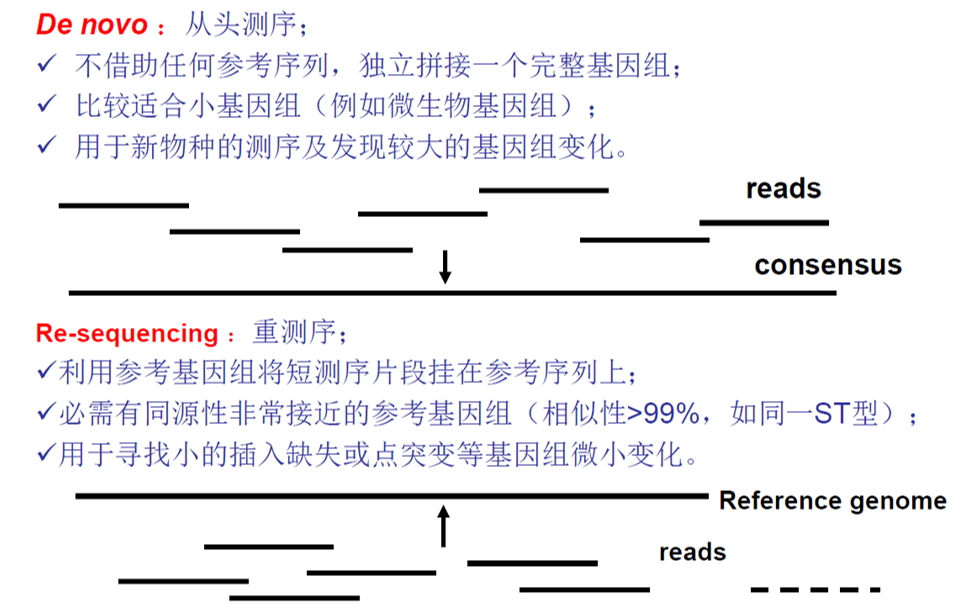
双末端测序 Pair-End、Mate-Pair
Mate-Pair 大片段环化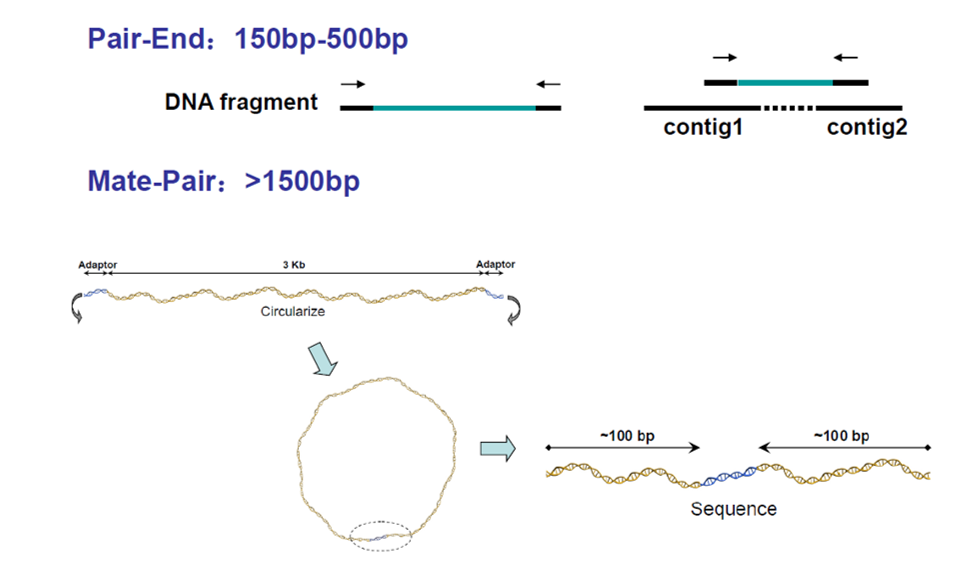
覆盖度、质量值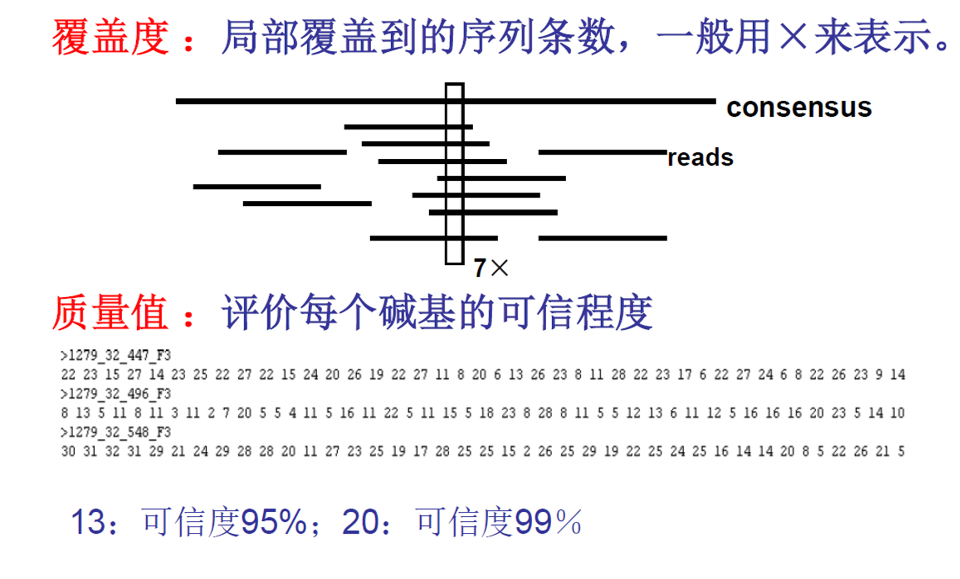
不同的芯片类型
- 表达谱芯片
- EST
- RNA-seq
- 其他芯片类型
表达谱芯片
单色荧光系统
- 较短的寡核苷酸芯片如 Affymatrix 芯片;载有较长片段的寡核苷酸芯片也可使用,如Illumina 芯片
- 实验组和对照组分别用同样的 荧光物质标记(或生物素biotin) ,分别与芯片杂交,即 一张芯片只杂交一个样本
根据杂交结果判断待测样品同芯片上哪个片段结合,通过与对照组的比较转化成基因表达的改变
双色荧光系统
- cDNA 芯片技术及载有较长片段的寡核昔酸芯片
- 实验组及对照组两种组织的 mRNA 在反转录成 cDNA 的过程中分别标记上 Cy3 和 Cy5 两种荧光,制备成探针,竞争性 地与芯片上的核酸片段进行杂交
- 两种波长的 激光扫描 读取竞争杂交的结果,通过计算机处理就能确定芯片上基因所结合探针的量,通过计算两种 荧光强度的比值 来判断两种组织中 基因表达是否有变化。
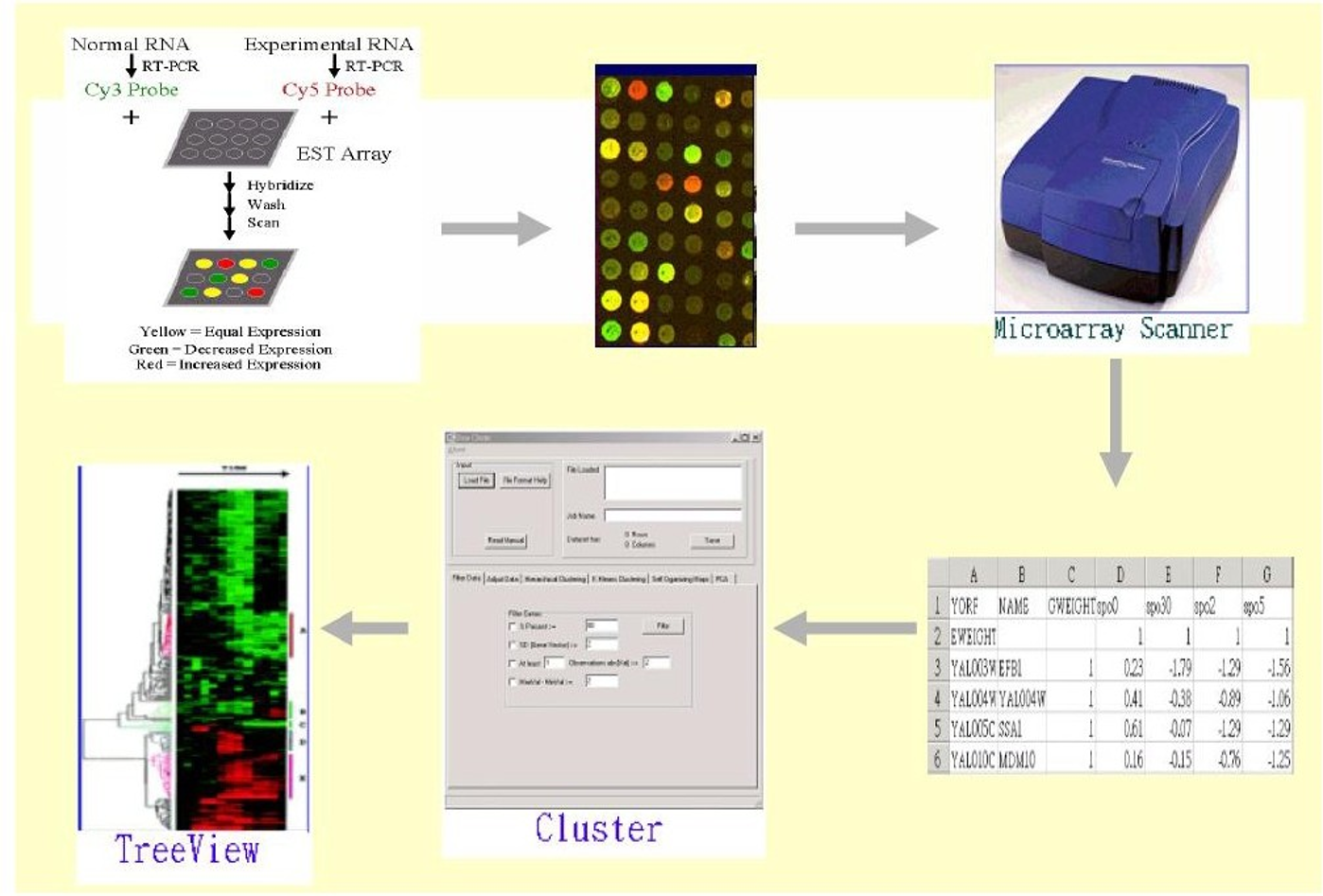
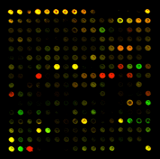
双荧光系统芯片的原理及流程图
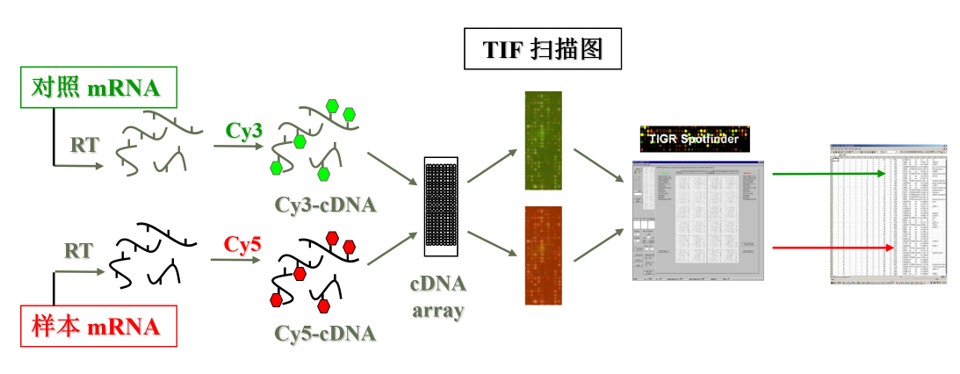
常见的双通道(dual channel)实验流程
- 对照基因(reference gene):绿色荧光标记(G)
- 样本基因(sample gene):红色荧光标记(R)
单通道和双通道的比较
单通道实验结果 重复性更好,使得比较不同样品之间各种基因表达的相对比例是可靠的。进行多个样品之间(芯片之间)的比较时,只需在多个样本中设置对照。由于点样的是寡核昔酸,探针无法检测。
双通道(点样cDNA)重复性相对较差,但是 探针可以测序验证。双色法需在每次杂交检测中设置对照,只能进行两个样品之间的比较,芯片之间的比较并不可靠
样本量:从多少总 RNA 中进行逆转录才能满足实验需要?
非扩增: 20 微克;扩增: 1 微克
有些情况下,样本极为稀有,不可能得到大量的 mRNA 靶分子
基因芯片实验对总RNA量的需求在一定程度上限制了该技术的推广
发展方向:提高检测灵敏度,减少基因芯片实验样本用量
数据分析
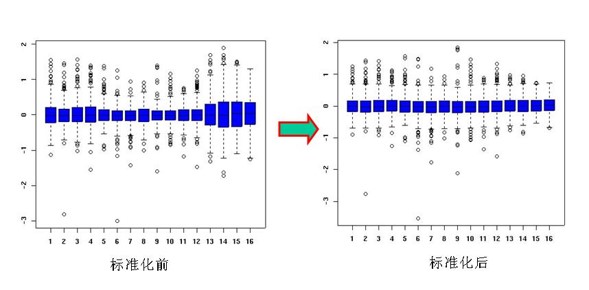
- 质控、均一化
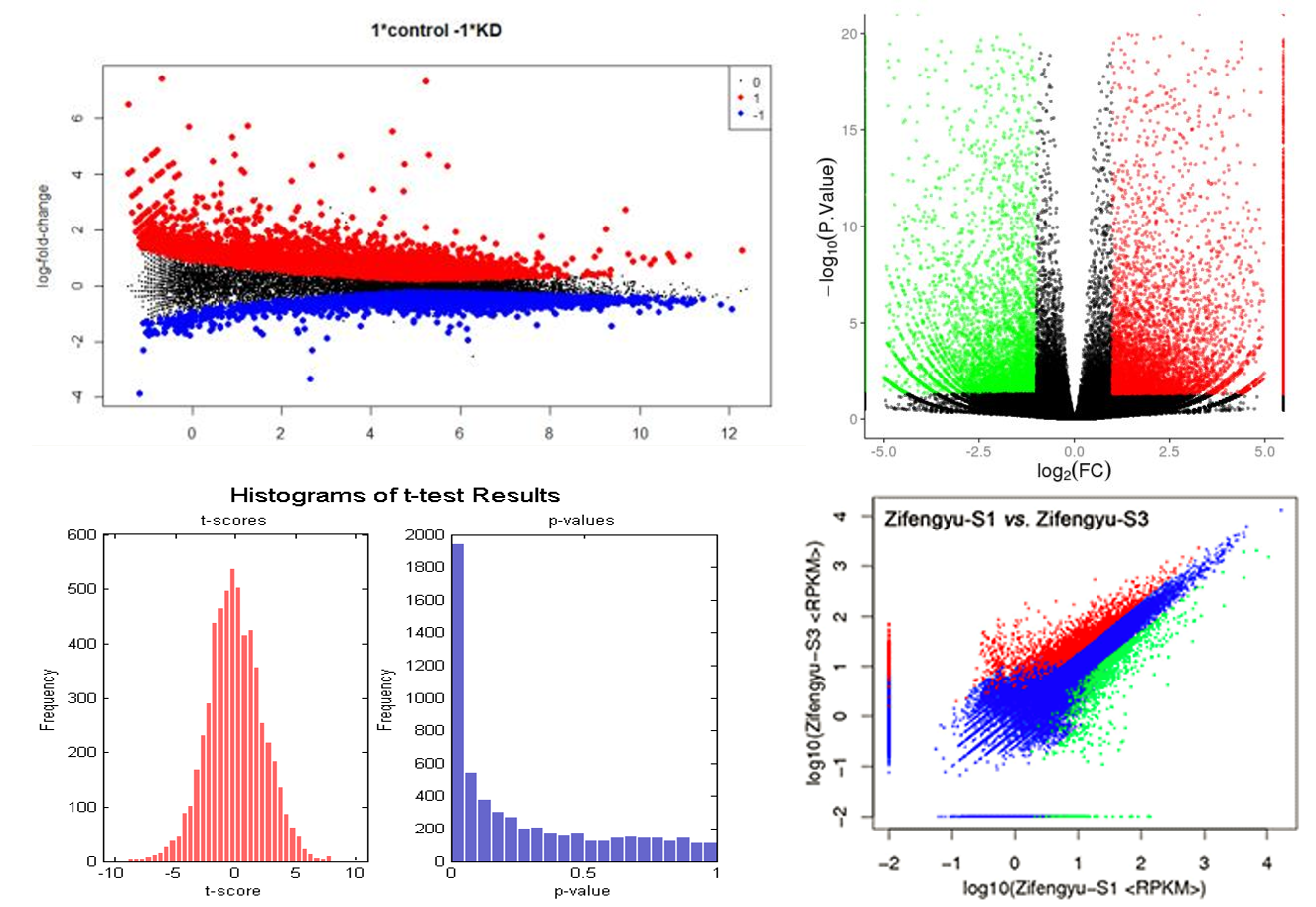
- 差异基因筛选
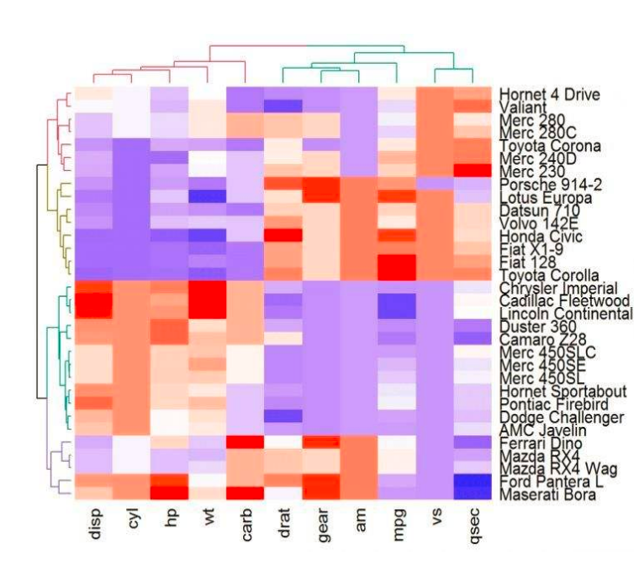
- 基因、样本聚类
- 功能富集分析
- 常用注释数据库:GO、KEGG、COG、Reactome、Biocarta等等;
- 功能富集分析方法主要分成四大类:
- ORA: over-representation analysis 过表达分析
- FCS:functional class scoring 功能集打分
- PT:pathway topology 通路拓扑结构
- NT:network topology 网络拓扑结构
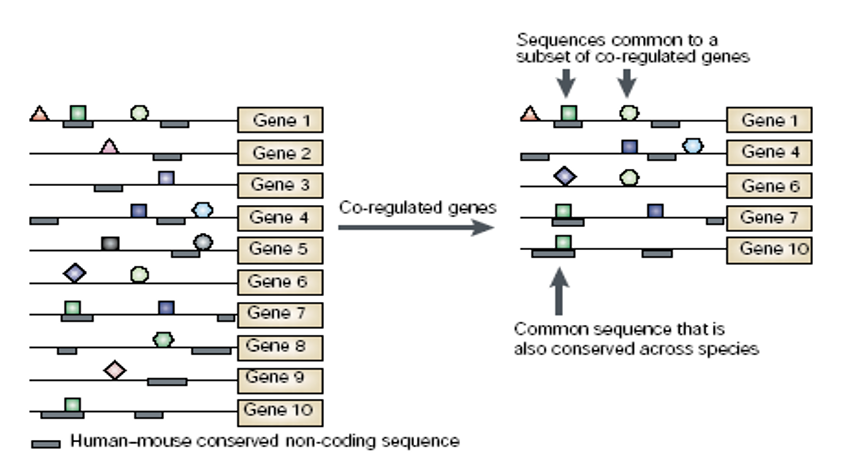
共调控基因
- 可能参与一些紧密相关的生理功能
- 可能由共同的转录因子激活
EST
ESTs(Expressed Sequence tags )是从 已建好的cDNA库 中随机取出一个克隆,从5’末端或3’末端对插入的 cDNA 片段进行一轮 单向自动测序,所获得的约60-500bp的一段cDNA序列。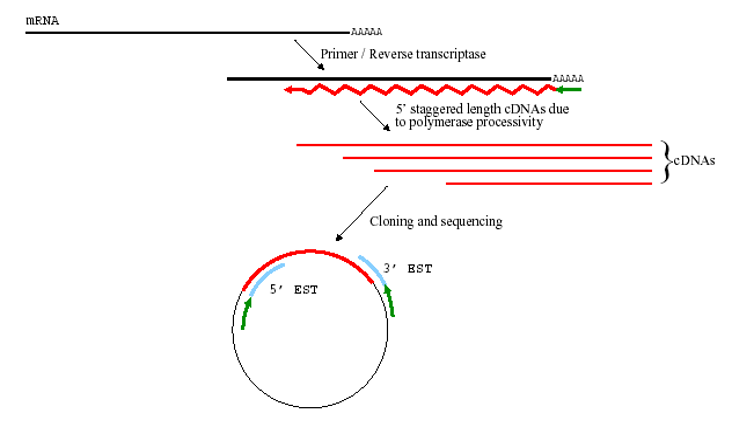
cDNA 文库分类
- Oligo d(T) cDNA文库。
- 随机引物cDNA文库。
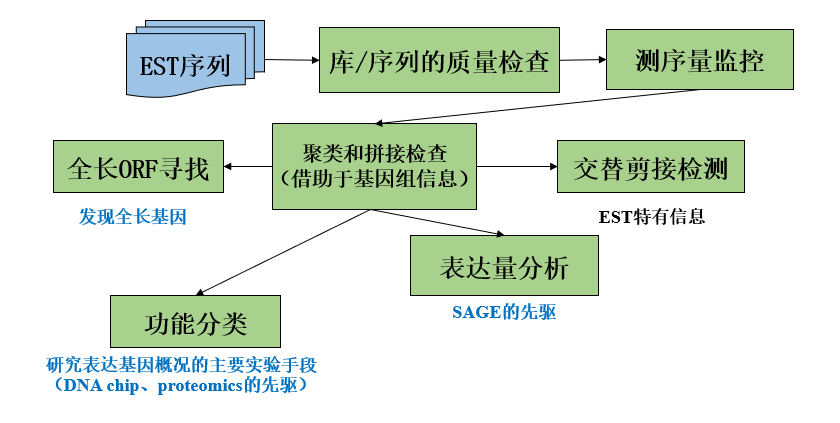
操作流程
- 随机挑取克隆进行5’或3’端测序
- 序列前处理
- 聚类和拼接
- 基因注释及功能分类
- 后续分析
序列前处理
- 应用 去除低质量 的序列 (phred)
- BLAST、RepeatMasker 或 Crossmatch 遮蔽数据组中 不属于表达的基因的赝象序列 (artifactual sequences)。
- 镶嵌克隆的识别
- 最后去除长度小于100bp的序列。
(序列长度:无统一标准,一般认为100bp以上的EST即可代表足够表达基因信息)
ESTs的聚类和拼接
- 聚类目的:将来自同一个基因或同一个转录本的具有重叠部分 (over-lapping) 的ESTs整合至单一的簇(cluster) 中。
- 聚类作用:
- 产生较长的一致性序列(consensus sequence),用于注释。
- 降低数据的冗余,纠正错误数据。
- 可以用于检测选择性剪切。
- 基因表达谱分析。
不严格的和严格的聚类
- loose clustering:
- 产生的一致性序列比较长
- 表达基因ESTs数据的覆盖率高
- 含有同一基因不同的转录形式,如各种选择性剪接体
- 每一类中可能包含旁系同源基因(paralogous expressed gene)的转录本
- 序列的保真度低
- stringent clustering:
- 产生的一致性序列比较短
- 表达基因ESTs数据的覆盖率低
- 因此所含有的同一基因的不同转录形式少
- 序列保真度高
基因注释及功能分类
- 基因注释:
- 序列联配:Blastn, Blastx
- 蛋白质功能域搜索 (结构比对):Pfam,Interproscan
- 基因功能分类:
- Gene Ontology分类
- COG分类
- KEGG分类
RNA-seq
- 样品RNA准备
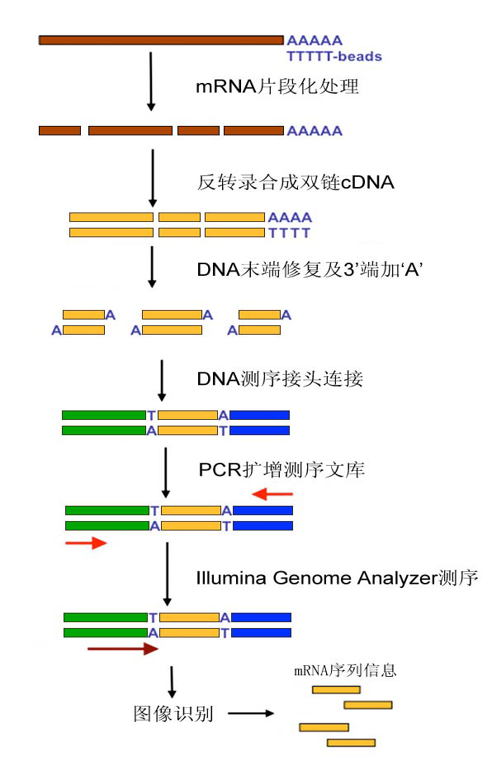
- 测序文库构建
- 使用 oligo dT 微珠纯化mRNA
- mRNA 片段化处理
- 反转录反应合成合成双链 cDNA
- 双链 DNA 末端修复 及 3’ 末端加 ‘A’
- 使用特定的 测序接头连接 DNA 片段两端
- 高保真聚合酶扩增构建成功的测序文库
- DNA成簇(Cluster)扩增
- 高通量测序(Illumina Genome Analyzer IIx)
- 数据分析
- 原始数据读取
- 与数据库比对并进行注释
- 深层次数据分析
fasta 和 fastq 格式
fastq
fasta

RNA-seq 数据的均一化
RPKM(Reads Per Kilobase per Million mapped reads),代表每百万reads中来自于某基因每千碱基长度的reads数。RPKM是将map到基因的read数除以map到基因组上的所有read数(以million为单位)与RNA的长度(以KB为单位)。
RPKM不仅对 测序深度 作了归一化,而且对 基因长度 也作了归一化,使得不同长度的基因在不同测序深度下得到的基因表达水平估计值具有了可比性,是目前最常用的基因表达估计方法。
技术优势
相对于传统的 sanger 测序法,RNA-seq具有以下优势:
- 数字化信号:直接测定每个转录本片段序列,单核苷酸分辨率的精确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题。
- 高灵敏度:能够检测到细胞中少至几个拷贝的稀有转录本。
- 任意物种的全基因组分析:无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析。同时能够检测未知基因,发现新的转录本,并精确地识别可变剪切位点及cSNP , UTR 区域。
技术应用
- 转录本结构研究:利用单碱基分辨率的 RNA-Seq 技术可极大地丰富基因注释的很多方面内容,包括 5’/3’边界鉴定、 UTRs 区域鉴定 以及 新的转录区域鉴定等。 RNA-seq 还可对 可变剪接( Alternative Splicing )进行 定量 研究。
- 转录本变异研究:在发现序列差异方面,如 融合基因 鉴定、编码序列多态性 研究等, RNA-seq 也具有很大的潜力。
- 非编码区域功能研究:转录组学研究的一个重要方面就是发现和分析ncRNA。ncRNA按其功能可分为 看家ncRNA 和 调节ncRNA。前者通常稳定表达,发挥着一系列对细胞存活至关重要的功能,主要包括转移RNA ( tRNA )、核糖体RNA ( rRNA )、小核 RNA ( snRNA ) 及小核仁 RNA ( snoRNA )等;后者主要包括长链 ncRNA ( IncRNA )和以 microRNA 为代表的小 snRNA( small ncRNA ) ,在表观遗传、转录及转录后等多个层面调控基因表达。
- 低丰度全新转录本的确定:虽然利用转座子标签和芯片技术能够获得全新的转录本,但是其工作量大,结果不确定。而 RNA-Seq 不受背景噪音问题的困扰,结果准确性高,因而被用来发现全新的转录本。近年来对酿酒酵母、栗酒裂殖酵母、拟南芥、水稻、小鼠、人、和人体白色念珠菌的转录组测定结果都显示出大量的新转录区域,并且其中许多转录水平都低于已知的cDNA 基因。
其它芯片
- 核酸水平
- 表达谱芯片
- CGH芯片
- 芯片测序
- MicroRNA芯片
- ChlP-chip(ChlP-seq)
- 非核酸水平
- 蛋白质芯片
- 组织芯片